

图片摘要
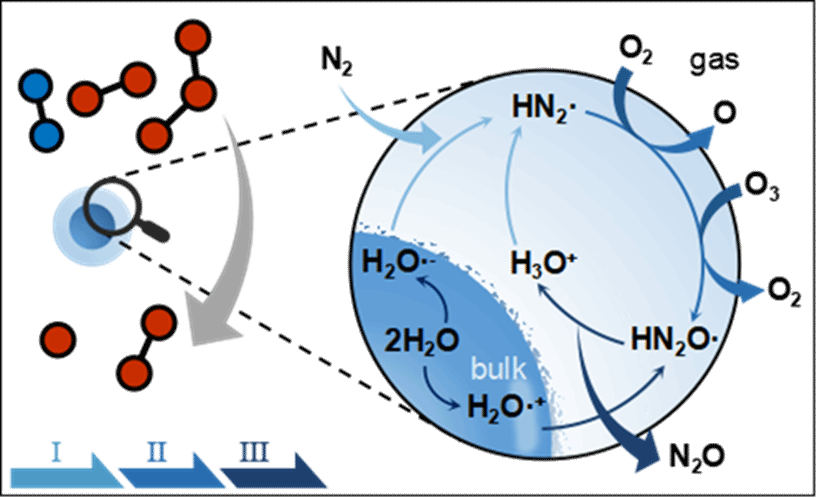
成果简介
近日,大连理工大学陈景文教授团队、宾夕法尼亚大学Joseph S. Francisco教授、哈尔滨工业大学姜杰教授团队合作在化学领域著名学术期刊Angew. Chem. Int. Ed.上发表了题为“Rapid N2O Formation from N2on Water Droplet Surfaces”的研究论文。该研究发现氮气(N2)能够在微液滴表面,通过独特的“先还原再氧化(RTO)”机制,快速转化成重要的温室气体和臭氧(O3)损耗物质氧化亚氮(N2O)。
引言
N2O是平流层O3损耗的重要贡献者,也是一种强效温室气体。自工业时期以来,N2O的浓度增加了大约20%,对平流层O3损耗和全球变暖的贡献日益增大。通常认为土壤和海洋中的微生物过程、农业和工业排放等人为活动是大气N2O的主要来源。大气中的二次途径(如在闪电的作用下N2生成N2+,进而生成N2O)被认为仅发挥较小的作用。
本研究揭示了一种在常温常压条件下,微液滴表面发生的RTO机制。该反应通过O2/O3迅速激活稳定的N≡N三键,导致大量N2O的形成(在实验反应器中可达ppmv·h-1量级)。根据实验结果的外推估计表明,RTO途径对全球N2O的贡献显著,远超过已知来源。
图文导读
N2O的自发生成现象
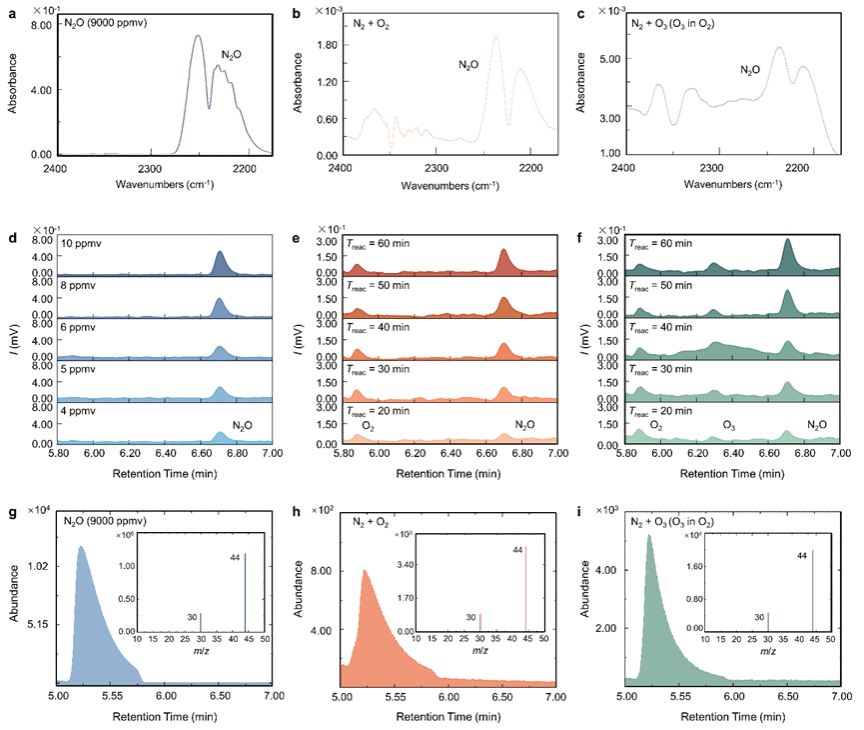
图1. N2和O2/O3混合气体在微液滴表面反应生成N2O的定性检测 [(a)-(c), 傅里叶变换红外光谱图;(d)-(f), 气相色谱图;(g)-(i), 气相色谱-质谱测定的总离子流图和质谱图]
在密封的玻璃反应器中,通过超声雾化方法持续生成直径为2 ~ 40 μm的液滴。在室温(20 ~ 25 ℃)和大气压力(1 atm)条件下,在反应器经过重复抽真空和充入氮气5次后,将4:1混合的N2和O2引入反应器,并开展微液滴实验。随后,分别通过傅里叶变换红外光谱仪(FTIR)、气相色谱仪(GC)和气相色谱-质谱联用仪(GC-MS)对收集的气态产物,进行定性和定量分析。
如图1a, b和c所示,在反应60 min后,FTIR中观察到了两个显著的峰,分别位于2238 cm-1和2214 cm-1。这些峰对应于N2O不对称伸缩模式的R和P支的耦合振动转动带。气相色谱结果表明(图1d, e和f),随着微液滴实验总反应时间的增加,N2O的浓度显著增加(保留时间约为6.75 min)。同时,GC-MS也检测到了N2O+ (m/z = 44)的母离子及其由N=N键断裂产生的NO+ (m/z = 30)的碎片离子(图1g, h和i)。值得指出的是,当加入一定浓度O3时,FTIR, GC和GC-MS信号更强,表明O3比O2更有效地促进N2O的形成。相较而言,在没有微液滴存在的条件下,FTIR, GC-ECD和GC-MS信号低于检出限。这些结果表明,微液滴显著促进了N2O的形成。
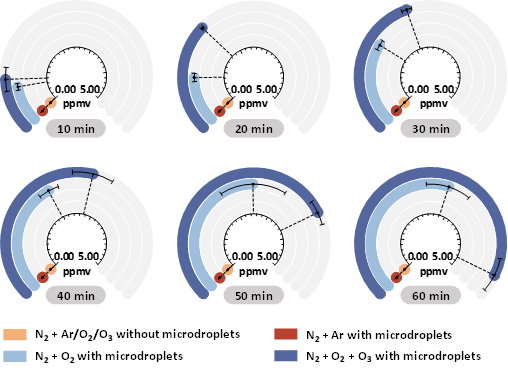
图2. 生成N2O的浓度随反应物组成(N2与其他气体的体积比为4:1;O2与O3的体积比约为0.98:0.02)和反应时间(Treac = 10, 20, 30, 40, 50, 60 min)的变化
N2O的自发生成机制
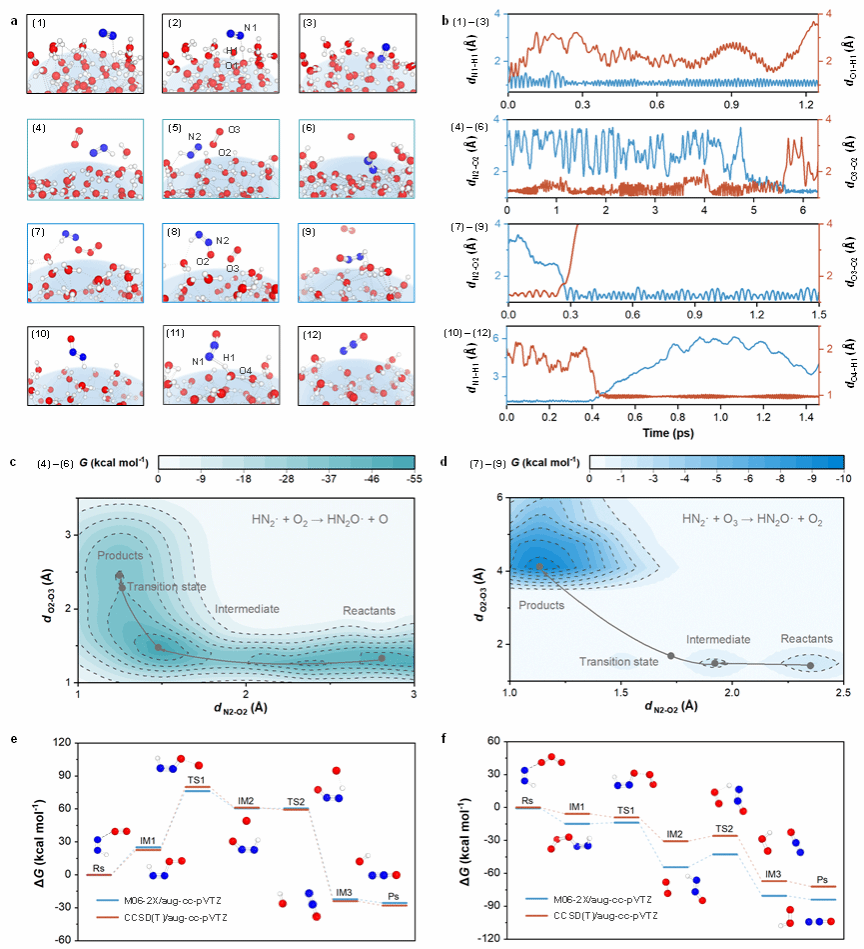
图3. N2O生成机理 [(a), 通过从头算分子动力学(AIMD)模拟获得的N2和O2/O3在微液滴表面反应的代表性瞬照;(b), AIMD模拟中观测到的键长随时间的变化;(c)和(d),O2和O3在液滴表面引发HN2·氧化的自由能变曲线;(e)和(f), 气相HN2·与O2和O3随后反应的代表性几何形状和自由能变曲线;红色、蓝色和白色的球体分别表示O, N和H原子]
如图3所示,通过传统分子动力学(MD)模拟、从头算分子动力学(AIMD)模拟、量子化学计算,揭示出了N2O在微液滴表面生成的机制。首先,MD模拟的结果表明,N2展现出了表面富集倾向。因此,N2有潜力在微液滴表面被活化并进一步被化学转化。
在0.5 ps的AIMD模拟中,直接观察到N2分子在H3O+存在的情况下被界面上的H2O·-自发还原(图3a和b)。该反应开始于H原子从H3O+转移到N2,随后电子从H2O·-转移到H3O+,生成关键中间体HN2·。形成的HN2·表现出显著的双N=N键性质,能在微液滴界面与O2或O3快速反应,生成“HN2O·+ O”或“HN2O·+ O2”。在此步骤中,形成一个长度为1.20 Å的N-O键,O2或O3中的O-O键逐渐拉长。后续由O2或O3氧化HN2·的自由能垒(ΔG‡) 分别为17.6或1.20 kcal mol-1 (图3c和3d)。AIMD结果表明,O3的参与可以进一步促进N2O的形成,与实验结果相符(图2)。随后,形成HN2O·和H2O·+在0.5 ps内经历一个无能垒的自由基-自由基复合反应,从而生成N2O。
由于形成的HN2·也可能挥发到气相中,本研究使用量子化学计算进一步研究了HN2·与O2/O3之间的气相反应。在M06-2X/aug-cc-pVTZ(图3e和f中蓝色线)和CCSD(T)/aug-cc-pVTZ//M06-2X/aug-cc-pVTZ(图3e和f中红色线)水平下,优化了几何构型并计算了自由能变。在O2参与的反应中,HN2·经历了“HN2· → HN2O2· → HN2O· → N2O”的路径,此过程具有极高的自由能垒(ΔG‡M06-2X= 51.2 kcal mol-1;ΔG‡CCSD(T)= 57.4 kcal mol-1);同时,气相O3参与反应的ΔG‡也高于微液滴表面反应的ΔG‡。因此,与在微液滴表面观察到的无能垒反应相比,气相反应在动力学上更加困难。综上,N2O主要在微液滴表面通过独特的RTO机制生成。
小结
本研究得到了国家自然科学基金(22206019, 22136001, 22074026)、博士后创新人才支持计划(BX20220050)、中央高校基本科研业务费(DUT24RC(3)047)、大连理工大学超级计算中心和曙光智算的支持。
作者简介
通讯作者:夏德铭,大连理工大学环境学院副教授,博士生导师。主要从事大气环境化学和环境计算化学等领域研究;发表论文27篇,包括第一/通讯作者Angew. Chem. Int. Ed. 1 篇、J. Am. Soc. Chem. 2篇、Environ. Sci. Technol. 3篇;主持博士后创新人才支持计划、青年科学基金等项目。
邮箱:xiadm@dlut.edu.cn。
文章链接:https://onlinelibrary.wiley.com/doi/abs/10.1002/ange.202421002