
研究亮点
• 阐明PVFE内部多孔网络结构抑制离子扩散,确保离子在电场方向的定向迁移。
• 构建仿真和数学模型强化对系统内离子迁移机理的理解并量化操作因素对离子迁移能力的影响。
• 指出过电流引发的H⁺和OH⁻共迁移是系统能耗损失的根本原因。
• 评估了PVFE处理实际高盐废水的技术和经济可行性。
研究进展
电化学分离技术被证实是实现高盐废水中酸/碱高效分离的有效手段,并具备运行条件温和、无需添加药剂的技术优势。本研究开发的制备型垂直自由流电泳(PVFE)技术,通过采用石英砂填料替代传统电分离技术中的膜组件,进一步克服了膜污染与高成本问题,在高盐废水资源化领域展现出显著优势。尽管该技术已成功应用于实际废水体系的酸/碱回收,但当前研究过度聚焦于运行参数优化,导致对系统内离子迁移机理的认知仍不清晰。因此,本研究以硫酸钠体系为研究对象,系统考察了电压、流速及进料浓度对PVFE内离子迁移行为的影响,深入解析其迁移驱动机制。指出石英砂填料可有效抑制体系内的离子扩散效应,同时保障离子在电迁移-对流耦合过程中保持定向迁移。此外,通过构建仿真模型与经验模型,不仅验证了离子传质机理,更量化了关键操作参数对离子迁移通量的调控规律,为PVFE系统的结构设计与性能强化提供了理论依据。
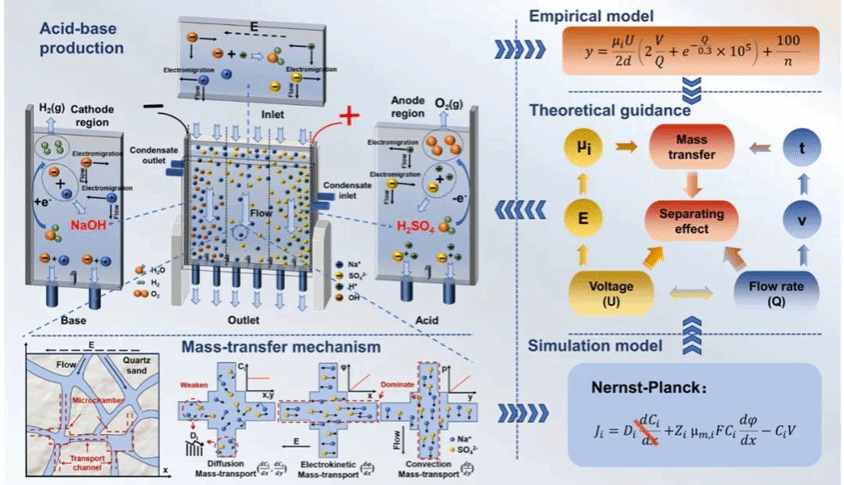
图1 图文摘要
为最大程度降低离子间相互干扰,精确反映不同电压与流速条件下 PVFE 中的离子迁移情况,初始阶段采用低浓度Na₂SO₄溶液作为进料。图2a–2e展示了PVFE中不同条件下的离子传质结果,从出口1(阴极侧)到出口6(阳极侧),Na⁺和SO₄²⁻存在显著的浓度梯度变化。Na⁺在阴极附近富集而在阳极附近贫化,SO₄²⁻的浓度变化与之相反。图 2f 所示的总质量平衡(M=1)证实系统处于稳态运行,无质量积累。如图 2g 所示,PVFE中石英砂填料堆积形成的多孔结构将整体分离室分割成众多微小空间单元,其中若干微室通过传输通道相互连接,最终构成多孔网络结构。运用微分思想可知,随着各单元空间尺度的减小,微室内部的浓度梯度显著趋缓(图2h)。另外,填料还提升了离子与孔壁的碰撞频率,导致扩散系数减小,从而显著削弱了微小单元间因扩散引起的物质传递。进而确保了Na⁺和 SO₄²⁻离子的有效电迁移和对流传质。
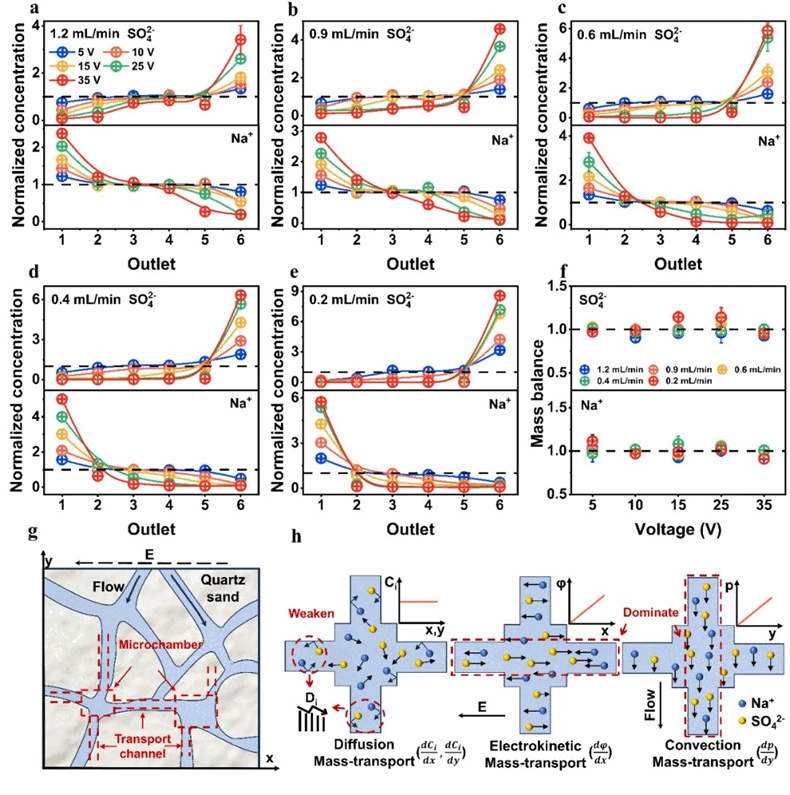
图2 (a–e)不同流速和施加电压下的离子迁移;(f)各离子组分的质量平衡;(g)石英砂形成的多孔介质示意图,展示离子与流体的传输通道;(h)PVFE系统中的传质机制,阐明电解质浓度梯度、电势差和压力差如何协同实现基于电荷的选择性离子分离。
通过对比不同填料填充状态下的离子迁移结果,并结合 COMSOL仿真模拟,为深入理解离子迁移机理提供了有力佐证。图3a展示了在无填充体系中,即使在高电压、低流速条件下,Na⁺和SO₄²⁻也未表现出定向迁移。定向迁移现象仅出现在填充体系中,且迁移程度随填料尺寸减小而增强。此外,通过逐步降低仿真模型中扩散系数(Di)值,模拟结果与不同填料粒径下的实验数据高度吻合。直接证明PVFE中致密填料网络通过限制离子扩散,对定向离子迁移起着关键调控作用。图3b和3c展示了两种不同操作条件下系统内的离子传质轨迹均朝电极方向倾斜向下,且倾斜程度随电压升高和流速降低而显著增强。这表明,一旦扩散干扰被最小化,水平方向的电迁移与垂直方向的对流传质共同作用,导致了离子倾斜向下的迁移轨迹。 进一步证实电迁移与对流是PVFE系统中离子的主要传输途径。最终,图3d-e展示了两种操作条件下,Na⁺和SO₄²⁻在系统出口处的模拟浓度分布均与实测值高度吻合,不仅验证了模型的准确性,同时也确认了对PVFE系统中离子传质机制的正确理解。另外,本研究还基于图3f–3i的数据构建了数学模型,旨在清晰揭示离子迁移能力与各项操作参数之间的定量对应关系。该数学模型清晰揭示了离子迁移行为同时受到施加电压和流速的影响,且二者存在交互作用。数学模型的分析结果也证实了离子在PVFE系统内保持匀速运动。总体来说,仿真和数学模型加强了离子传质分析的有效性,可为PVFE系统的实际应用提供理论指导。
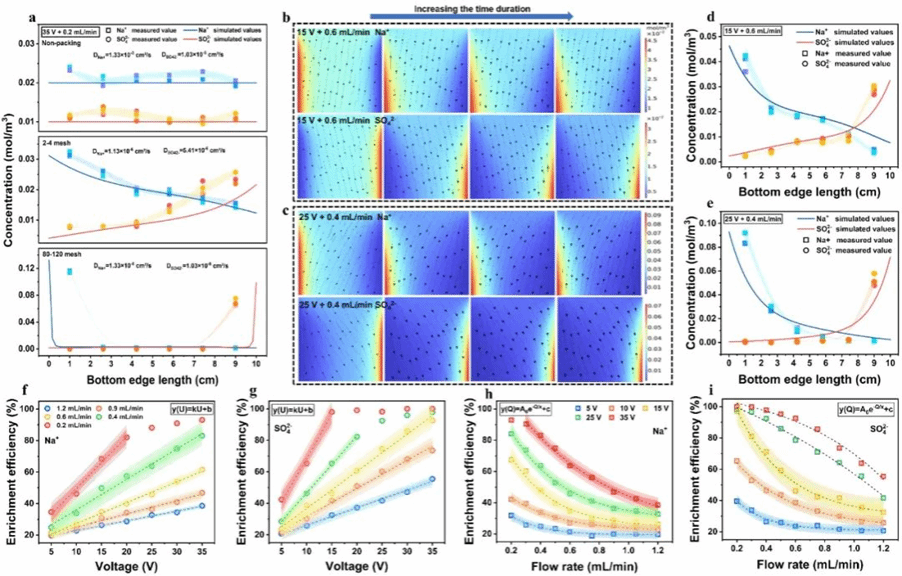
图3 (a)扩散系数与填料尺寸对出口离子浓度分布的影响;(b-c)15 V与0.6 mL/min及25 V与0.4 mL/min条件下PVFE体系中Na⁺和SO₄²⁻的动态分布;(d-e)Co₃O₄/CF,模拟与实测出口离子浓度分布对比;(f-g)施加电压对Na⁺与SO₄²⁻富集的影响;(h-i)进料流速对Na⁺与SO₄²⁻富集的影响
为评估PVFE体系中不同浓度下离子迁移行为的一致性,本实验采用浓度递增的Na₂SO₄溶液进行测试。结果表明无论进料浓度如何,两电极间始终维持显著的Na⁺与SO₄²⁻浓度梯度,且装置内未观察到物质积累现象(图4a、4b)。然而,在恒定电压与流速条件下,随着离子浓度升高,两极间浓度梯度逐渐减小。高浓度条件下,离子迁移行为对电压与流速变化的敏感性增强,其中SO₄²⁻的敏感性更为显著(图4c)。基于科尔劳施经验公式及离子缔合理论(图4f),上述现象可归因于高浓度下离子缔合作用增强,导致局部静电荷被部分中和。仿真与数学模型为预测不同浓度条件下PVFE体系内的离子浓度分布及电极附近的离子富集效率提供了直接方法。如图4d和4e所示,高浓度下的动态离子分布与低浓度下观测结果高度吻合,且实测出口浓度值与模拟值匹配良好。此外,图4c展示了数学模型拟合优度的评估结果,Na⁺与SO₄²⁻的R²分别为0.9685和0.9452。这些结果表明,在低浓度下建立的离子迁移模式,即使在离子浓度升高时仍保持一致。尽管较高的离子浓度可能增强离子间相互作用并影响电化学活性,但并未根本性改变离子在PVFE体系中的迁移行为。
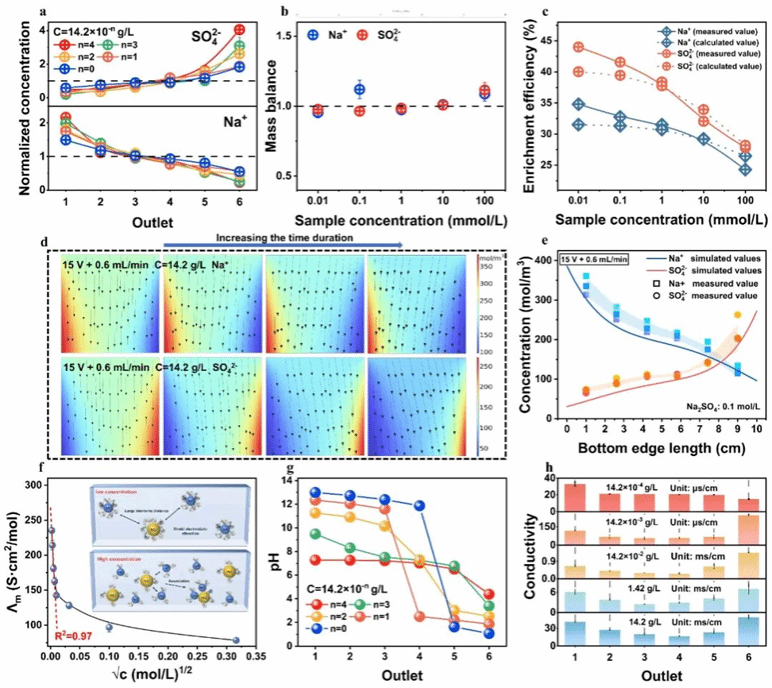
图4 (a-b)特定离子组分的传质与物料平衡;(c)电极附近Na⁺与SO₄²⁻的富集效率;(d)PVFE体系中Na⁺与SO₄²⁻的动态离子分布;(e)模拟结果与实测结果的出口离子浓度分布对比;(f)Na₂SO₄摩尔电导率(Ʌm)随浓度平方根(√c)的变化规律:在稀Na₂SO₄溶液中,离子以单分散水合离子形式存在,此时Ʌm与√c呈线性相关;而浓度升高则引发非线性偏离,此现象源于离子对的形成;(g-h)出口1至6间pH与电导率的变化情况
提高施加电压或降低流速均会导致能耗增加及电流效率下降(图5a)。该现象主要由两个因素导致:(1)根据第二维恩效应(图5f),高电压会加速水解离生成H⁺和OH⁻;(2)“隧道效应”(图5g)通过质子传导机制增强了H⁺和OH⁻的电荷传输。因此,在低进料浓度条件下,施加较高电压或采用较低流速会显著增加H⁺和OH⁻的生成与传输能耗,该部分能耗在不同工况下占总能耗的22.79%至67.24%(图5d)。由此可见,H⁺和OH⁻迁移比例的提升是系统能耗急剧增加的主导因素。图5b表明,提升系统分离性能会导致能耗增加。然而,在5 V/0.2 mL/min、10 V/0.6 mL/min 及 15 V/0.9 mL/min 的操作条件下,盐分离转化率稳定维持在~40%。但5 V的最低电压设置使能耗较其他工况降低了40%和52%,尽管此时流量条件不利于降低系统能耗。这一结果表明:外加电压是影响PVFE系统能耗的主导因素。此外,图5c表明高电压会因副反应增加导致电流效率下降。这些非目标反应造成额外能量损失,使得盐分离效率与能耗之间呈现需权衡的平衡关系。在相同条件下,提高离子浓度可降低能耗并提升电流效率(图5e)。最终,随着离子浓度升高,如图5h所示,阳极与阴极的电化学反应逐渐转向消耗H⁺和OH⁻。这源于水电解产生的H⁺与OH⁻同体系中存在的Na⁺和SO₄²⁻在数量上的失衡。由此,在PVFE体系中,H⁺和OH⁻离子对电流产生的相对贡献度降低。
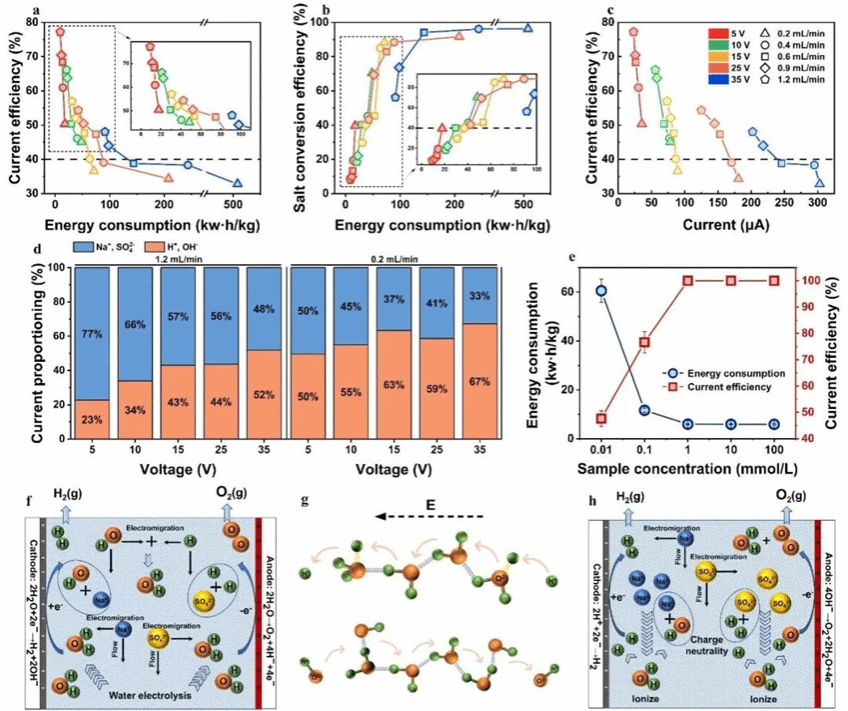
图5 (a-c)电流效率、盐分离转化率与能耗的相互关系;(d)不同工况下电流贡献比例;(e)不同浓度下能耗与电流效率;(f)低浓度下电极反应与离子传质过程;(g)H⁺与OH⁻的传质机制:氢键作用与水分子振动促进迁移;(h)高浓度下电极反应与离子传质过程
本研究深入阐明了PVFE内部离子传质机理,并通过仿真模型与数学模型相结合的手段证实了系统内石英砂填料堆积构建的多孔网络结构有效限制离子扩散,确保Na⁺与SO₄²⁻在电迁移和对流过程中保持正确定向,从而实现有效分离。另外,与传统膜分离技术不同,PVFE系统无需昂贵的膜结构,显著降低投入成本,尤为适用于小型工业废水处理。
